- FR
- EN
Aqueous corrosion
The corrosion of metallic materials at ambient or relatively low temperature is mainly due to liquid water coming to their contact. The aqueous phase can be neutral, acid or basic. Aqueous corrosion is an electrochemical process with origins in the characteristics of electrical conduction of the two phases: electronic conduction in the metal phase and ionic conduction in the aqueous phase, also called electrolyte. The electrochemical reactions enable the charge transfers on the interface between metal (electrode) and the electrolyte.
On certain zones of the interface metal - electrolyte, the metal get oxidized to form positively charged ions (or cations) that go in solution in the liquid or they can form solid compounds such as oxides, which remain on metal.
The reaction of the metal oxidation (anodic reaction) can be written as shown below
where M is the metal, Mz+ is the metallic cation with valence z.
This reaction of oxidation releases electrons which must be consumed to ensure electrical neutrality. This is why one or more reactions of reduction of oxidizing chemical species (cathodic reactions) present in the aqueous phase necessarily happens simultaneously at the interface. The most common cathodic reactions are the reductions of dissolved oxygen of H 3 O + ions or water:
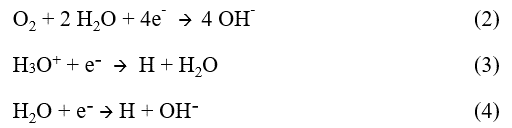
This process constitutes a pile of corrosion in which the current known as anodic goes from the metal towards the aqueous phase to the places where it corrodes (anodic zones) and the current known as cathodic enters metal where the oxidizing species are reduced (cathodic zones). If there is no external electrical current, the anodic and cathodic currents are equal and flowing in opposite directions.
The products of the reactions (1) and (2) are ions which can remain in solution or react to give a precipitate (corrosion product) according to the reaction:
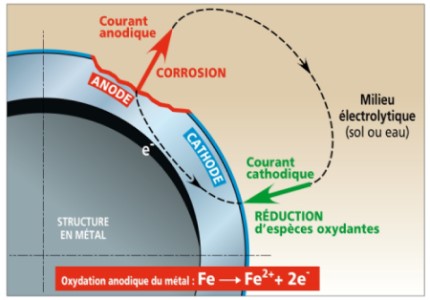
The pile of corrosion
In the case of ordinary steels, rust is formed. In certain cases (stainless steels for example) a very thin protective oxide coating is formed (some nanometres): then we talk about " passivity " and the residual corrosion rate is very low. Certain metals or alloys such as those made of aluminium or titanium, because of their high reduction power, oxidize quickly and get coated with a protective oxide layer.
In addition, the atoms of hydrogen formed on the cathodic zones tend to recombine in diatomic gaseous hydrogen:
However, part of this atomic hydrogen can penetrate in the metallic phase (especially in carbon steels) and lead to various forms of hydrogen embrittlement (in particular in the case of cathodic over-protection).
The corrosion rate of a metal is thus directly related to the intensity of the anodic and cathodic reactions that act at the interface. The flow of electrons, which is the corrosion current, is linked, by the Faraday's law, to the quantity of oxidized metal.
So, a metal corrosion rate can be expressed in various ways: in corrosion current (for example in microamperes per cm 2 , or in mass of oxidized metal per unit of time and per unit of area (for example mg per dm 2 and per day) or in thickness of oxidized metal per unit of time (for example µm per year).
For ordinary steel there is approximately the following correspondence:
1 µA/cm2 ≡ 1 µm/month ≡ 2,5 mg per dm2 and per day
or 1 mm/year ≡ 7,8 kg/m2.year ≡ 860 mA/m2
The significant parameters which are involved in the corrosion rate are numerous and depend on the material (composition, surface quality, metallurgical state...), on the environment (chemical composition, in particular pH and redox potential, complexants, etc...) and on the conditions at the interface (fluid flow, temperature...).
The kinetic approach of the study of corrosion by the oscillations of the diagrams current/potential density (polarization curves) enables to quantify the current exchanges and thus the corrosion rate and the need for cathodic protection current. These curves, using a D.C.generator (potentiostat) enable to determine the anodic reactions rate (metal oxidation) and cathodic (reduction of the environment oxidant – which can be water itself -) according to the electrode potential (measured with regard to a reference electrode –see below). If there is no external impressed current, the metal is at the free corrosion potential, the anodic and cathodic currents are equal in absolute value.
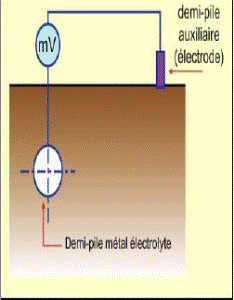
Measure of potential of free corrosion of a metal in an électrolyte
Diagram potential/pH (diagram of POURBAIX) for iron
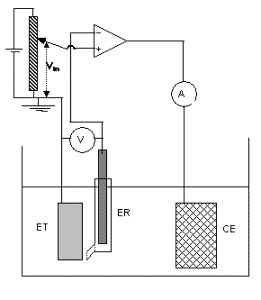
Layout of the polarization curves with a potentiostat
The potentiostat imposes a given value V in potential of the work electrode ET (metal) with regard to a reference electrode ER. This polarization of metal is obtained by the passage of a current, for which we measure the intensity I, enters ET and an auxiliary electrode (or counter electrode) CE.
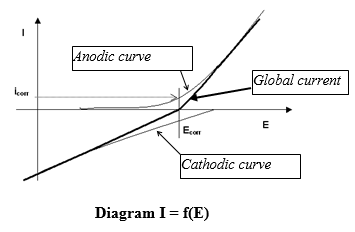
At the free potential of corrosion E corr the value of the anodic current (equal to the cathodic current) represents the corrosion current I corr. The total current measured in the working electrode /counter electrode circuit is the algebraic sum of the anodic and cathodic currents.
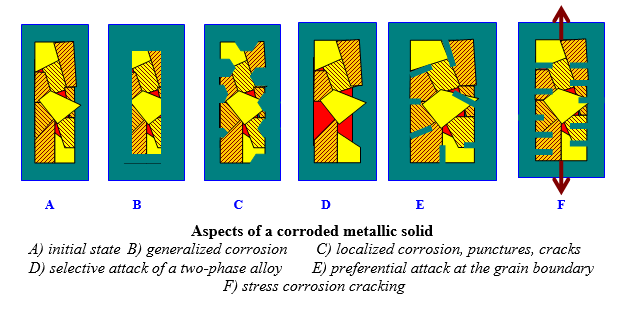
The electrochemical reactions are not distributed uniformly on the metallic surface. For example, the anodic zone (oxidation of metal) can have very variable localizations and size, characterizing the corrosion aspect (generalized corrosion, more or less uniform, or localised corrosion leading for example to the formation of punctures or cracks).
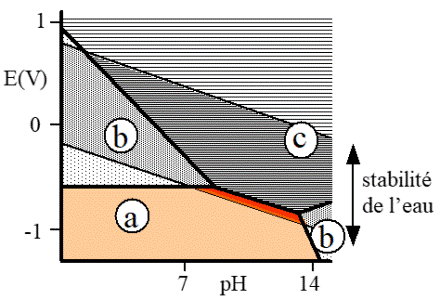
Potential/pH diagram (POURBAIX diagram)for iron
The immunity (a) corresponds if the metallic material is the stable species. The field (b) corresponds to a passage in solution of the metal ions. The zone (c) corresponds to the precipitation of a compound, on the metal. The field of stability of water is limited by the reactions of oxidation or reduction of water (formation of oxygen or hydrogen)
Thermodynamics enables to envisage the stability fields of the chemical species present in a system of corrosion. The potential pH diagrams (Pourbaix diagrams) obtained are precious auxiliaries to know the influence of a pH or potential variation on the stability fields or the prevalence of ions or considered compounds.
