- FR
- EN
La corrosion aqueuse
La corrosion des matériaux métalliques à température ambiante ou relativement peu élevée est essentiellement due à l’eau liquide venant à leur contact. La phase aqueuse peut être neutre, acide ou basique. La corrosion aqueuse est un processus électrochimique qui voit son origine dans le caractère de conduction électrique des deux phases en présence : conduction électronique dans la phase métallique et conduction ionique dans la phase aqueuse, appelée aussi électrolyte. Les réactions électrochimiques permettent les transferts de charge au niveau de l’interface entre le métal (électrode) et l’électrolyte.
Sur certaines zones de l’interface métal – électrolyte, le métal s’oxyde sous la forme d’ions chargés positivement (ou cations) qui passent en solution dans le liquide ou sous forme de composés solides tels que des oxydes qui restent sur le métal.
La réaction d’oxydation du métal (réaction anodique) peut s’écrire ainsi :
(1)
où M représente le métal, Mz+ est le cation métallique à la valence z.
Cette réaction d’oxydation libère des électrons qui doivent être consommés pour assurer la neutralité électrique. C’est pourquoi une ou plusieurs réactions de réduction d’espèces chimiques oxydantes (réactions cathodiques) présentes dans la phase aqueuse ont nécessairement lieu simultanément à l’interface. Les réactions cathodiques les plus courantes sont les réductions de l’oxygène dissous, des ions H3O+ ou de l’eau :
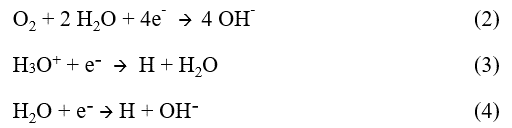
Ce processus constitue une pile de corrosion dans laquelle le courant dit anodique va du métal vers la phase aqueuse aux endroits où il se corrode (zones anodiques) et le courant dit cathodique entre dans le métal aux endroits où le ou les espèces oxydantes se réduisent (zones cathodiques ). En l’absence de courant électrique extérieur les courants anodique et cathodique sont égaux et de sens opposés.
Les produits des réactions (1) et (2) sont des ions qui peuvent rester en solution ou réagir pour donner un précipité (produit de corrosion) selon la réaction :
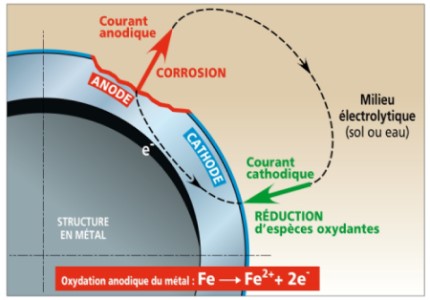
La pile de corrosion
Dans le cas des aciers ordinaires, on forme ainsi de la rouille. Dans certains cas (aciers inoxydables par exemple) il se forme une couche d’oxyde protectrice de très faible épaisseur (quelques nanomètres) : on parle alors de « passivité » et la vitesse de corrosion résiduelle est très faible. Certains métaux ou alliages tels que ceux d’aluminium ou de titane du fait de leur grand pouvoir réducteur s’oxydent rapidement et se recouvrent d’une couche d’oxyde protectrice.Par ailleurs, les atomes d’hydrogène formés sur les zones cathodiques ont tendance à se recombiner en hydrogène gazeux diatomique :
Cependant, une partie de cet hydrogène atomique peut pénétrer dans la phase métallique (notamment dans les aciers au carbone) et conduire à différentes formes de fragilisation par l’hydrogène (notamment en cas de surprotection cathodique).
La vitesse de corrosion d’un métal est donc directement liée à l’intensité des réactions anodiques et cathodiques qui se déroulent à l’interface. Le flux d’électrons, qui est donc le courant de corrosion, est lié, par la loi de Faraday, à la quantité de métal oxydé.
De ce fait la vitesse de corrosion d’un métal peut s’exprimer de diverses manières : en courant de corrosion (par exemple en microampères par cm²), ou en masse de métal oxydée par unité de temps et par unité de surface (par exemple mg par dm² et par jour) ou encore en épaisseur de métal oxydée par unité de temps (par exemple µm par an).
Pour l’acier ordinaire on a sensiblement la correspondance suivante :
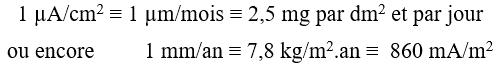
Les paramètres importants qui régissent la vitesse de corrosion sont nombreux et dépendent du matériau (composition, état de surface, état métallurgique…), du milieu (composition chimique, notamment pH et potentiel redox, complexants, etc…) et des conditions à l’interface (vitesse du fluide, température, …).
L’approche cinétique de l’étude de la corrosion par le tracé des diagrammes densité de courant/potentiel (courbes de polarisation) permet de quantifier les échanges de courant et donc la vitesse de corrosion et le besoin en courant de protection cathodique. Ces courbes, utilisant un générateur de courant continu (potentiostat) permettent de déterminer la vitesse des réactions anodique (oxydation du métal) et cathodique (réduction de l’oxydant du milieu –qui peut être l’eau elle-même-) en fonction du potentiel d’électrode (mesuré par rapport à une électrode de référence –voir ci-après). En l’absence de courant imposé extérieur le métal est à son potentiel de corrosion libre, les courants anodiques et cathodiques étant égaux en valeur absolue.
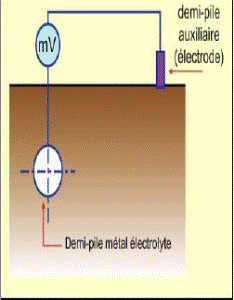
Mesure du potentiel de corrosion libre d’un métal dans un électrolyte
Schéma d'un diagramme potentiel/pH (diagramme de POURBAIX) pour le fer
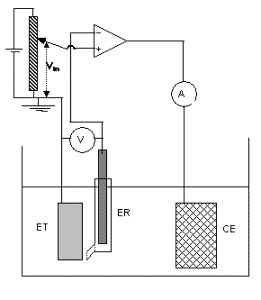
Tracé des courbes de polarisation au moyen d'un potentiostat
Le potentiostat impose une valeur déterminée Vin potentiel de l’électrode de travail ET (métal) par rapport à une électrode de référence ER. Cette polarisation du métal est obtenue par le passage d’un courant, dont on mesure l’intensité I, entre ET et une électrode auxiliaire (ou contre-électrode) CE.
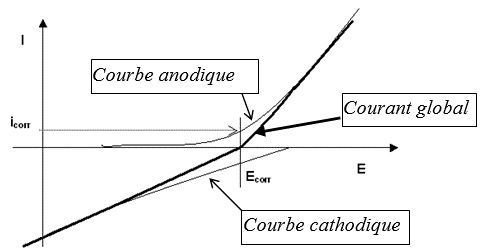
Schéma d'un diagramme I=(f)E
Au potentiel libre de corrosion Ecorr, la valeur du courant anodique (égale au courant cathodique) représente le courant de corrosion icorr. Le courant global mesuré dans le circuit électrode de travail/contre-électrode est la somme algébrique des courants anodique et cathodique.
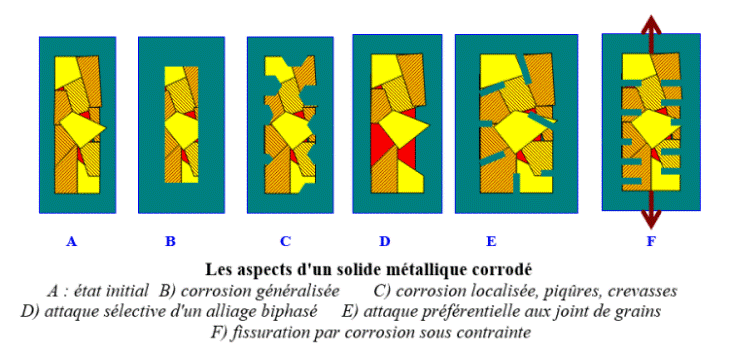
Les réactions électrochimiques ne sont pas distribuées de façon uniforme en tout point de la surface métallique. Par exemple, la zone anodique (oxydation du métal) peut présenter des localisations et étendues très variables, caractérisant l’aspect de la corrosion (corrosion généralisée, plus ou moins uniforme, ou corrosion localisée conduisant par exemple à la formation de piqûres ou de fissures).
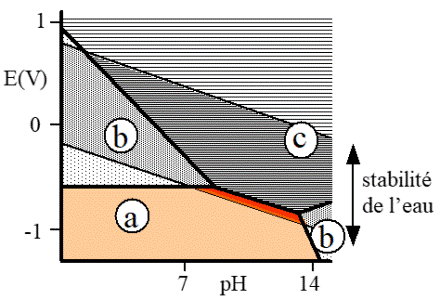
Shéma d'un diagramme potentiel/pH (diagramme de POURBAIX) pour le fer
La thermodynamique permet de prévoir les domaines de stabilité des espèces chimiques présentes dans un système de corrosion. Les diagrammes potentiel pH (diagrammes de Pourbaix), ainsi obtenus sont des auxiliaires précieux pour connaître l’influence d’une variation de pH ou de potentiel sur les domaines de stabilité ou de prédominance des ions ou composés considérés.
La zone (a) correspond au domaine d’immunité dans le cas où le matériau métallique est l’espèce stable.
La zone (b) correspond à un passage en solution des ions métalliques.
La zone (c) correspond à la précipitation d’un composé sur le métal.
Le domaine de stabilité de l’eau est limité par les réactions d’oxydation ou de réduction de l’eau (formation d’oxygène ou d’hydrogène).
